一、对接完整步骤
蛋白质:去水,加全氢,导出为PDBQT文件格式(设置为受体)
小分子:加全氢,设置为配体(自动分布电荷),检测扭动键,选择扭动键,导出为PDBQT
设置对接box,导出为GFP,运行Autogrid4,设置对接参数及运算方法,运行Autodock,查看结果
二、具体的操作步骤
关于蛋白的处理
1.设置对接的目录,就是之前的这三个文件所在的位置就是我的对接的目录,据我而言,一般就是哪有就有了。


2.然后就是将你需要对接的小分子和蛋白质之类的全部放在这个目录里面,相当于这个就是我们对接用的目录文件夹了。

3.导入蛋白质,File——read molecules,然后双击蛋白质,导入以后我们会发现这里会有很多的小红点,这些小红点代表的水分子,现在我们就是需要将这些水分子进行去掉。然后我们就需要Edit——Delete Water去除水分子。


4.接下来就是进行加氢操作,Edit——Hydrogens——Add,然后弹出来的那个你点击ok就可以。下面那张图片是加完氢以后的图片,就是增加了很多的小白点。


5.接下来就是将我们的蛋白质选择为受体,Grid——Macromolecules——choose——选择我们的蛋白质——Select Molecules,然后等待一会会弹出一个报错,不要管,点击确定即可。然后确定完了以后,它会直接蹦到你的工作目录了,然后命名啥的也不用管,你就直接确定就好了。

6.到此为止关于蛋白的处理就已经结束了,然后找到我们左边的这个选择这个蛋白质,然后删除这个蛋白质,开始对我们的小分子进行处理。

关于小分子的处理
1.导入小分子,File——read molecules,然后双击小分子,小分子的格式可以是PDB,也可以是mol2文件。
2.然后给小分子进行加氢处理,Edit——Hydrogens——Add,然后弹出来的那个你点击ok就可以。
3.接下来将小分子选择为配体,Ligand——Input——Choose,然后弹出以后选择你刚刚的那个小分子,然后完了以后会弹出一个报错,不要紧

4.接着我们检查一下这个小分子的中心,Ligand——Torsion——Delect Root,完了之后会出现一些小球球。

5.然后接下来,我们检测一下小分子的扭转键,Ligand——Torsion Tree——Choose Torsions,然后红色的是可以扭转的键,然后绿色的是可以扭转的键。完了之后我们点击Dnoe


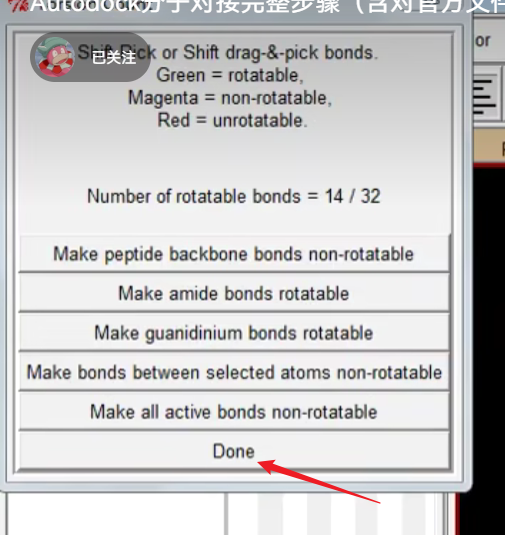
6.接下来我们就是将我们的小分子导出为PDBQT文件,Ligand——Output——Save as PDBQT,然后同样的会弹出到我们的工作目录中,然后也是什么都不用管,直接按回车就可以。

7.接下来就是删除这个小分子,操作过程和前面是一样的,然后我们接下来就是准备开始分子对接了。
分子对接
1.接着开始进行分子对接,Grid——Macromolecules——open,然后就是双击蛋白质的PDBQT文件。弹出弹框后点击yes。

2.接下来就是导入小分子,Grid——Set Map——open Ligand,然后就是点击小分子的PDBQT文件

3.然后为了方便演示,这里就给小分子换一个模型,就是让它们更加明显。
4.然后就是需要准备一个分子对接的box,Grid——Grid box。

6.然后就是调整这个box,使它们包含这个蛋白质,当然了如果是知道活性位点的话,我们也可以只包括活性位点就可以了。这个上面这三个是调整大小的,然后0.375那个是调整box的比例的,然后下面三个是移动位置的。

7.然后我们需要将我们的配体从box中取出来,点击root——选中小分子——取消那个mouse,然后我们右键选中小分子,然后将其拖出我们的box,然后就是继续在这个界面中我们再勾选上那个mouse的勾勾,然后关闭这个界面就可以了。


8.接下来保存现在盒子的样子,file——close saving current。

9.然后就是导出GPF文件,Grid——Output——save GPF,然后这里需要我们主动给这个文件命名,然后一般的命名都是Grid.gpf,注意命名中一定是要以.gpf结尾才可以。

10.然后运行Autodock,Run——Run AutoGrid,然后就会弹出下面这个框,然后就是点击第三个Browse,然后双击刚才的gpf文件,然后什么都不用管,直接点击launch。


11.然后就是蹦出了一个小框,还有就是工作目录出现很多map文件,静静等待小方框消失即可。

12.然后就是设置对接的一些参数了,选择Docking——Macromolecules——set Rigid Filename,这里因为我们做的是半柔性对接,所以不选择下面那个柔性对接。然后就是选择蛋白质的PDBQT文件,然后双击它。然后Docking——Ligand——choose,选择小分子。

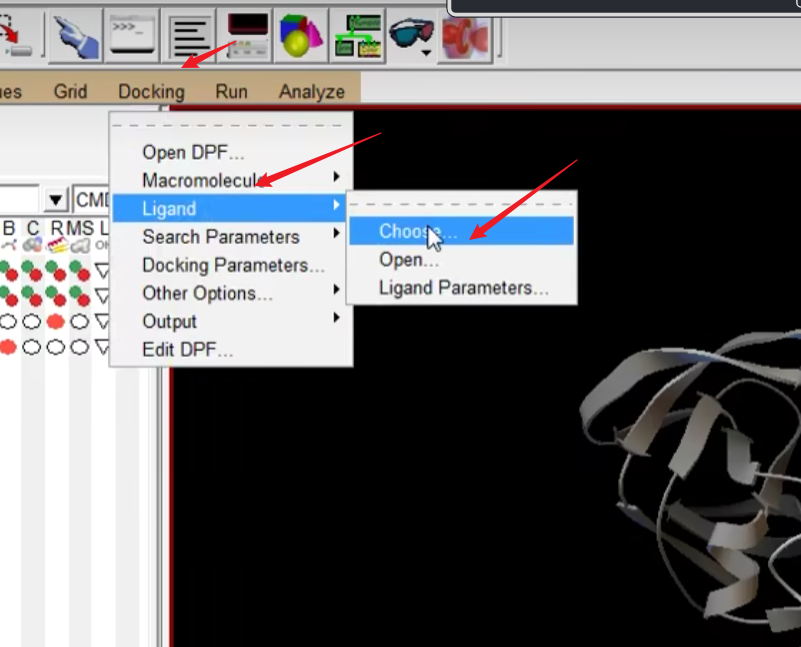
13.然后Docking——Search Parameters——Genetic Algorithm,然后弹出来的东西选择接受。然后选择Docking——Docking Parameters,然后一连弹出很多框,只需要最后那个点选择接受就行。然后就是Docking——output——Lamarckian,然后这里我们需要手动输入一个后缀名为dpf的文件,然后一般输入dock.dpf文件即可。



14.接下来运行Autodock,Run——run autodock,然后就蹦出来一个和前面那个一样的界面,然后同样也是我们只需要管第三个browse,然后选择刚刚那个dock.dpf文件就可以了。然后什么都不管,点击lanuch就可以了。然后我们的工作目录就多了一个dlg文件。
15.接着我们就是删除所有的分子,准备查看对接结果。

16.然后就是Analyze——Docking——open,双击刚刚的dlg文件,然后就是会蹦出来一个界面,就是说有10个对接状态,然后点击确定就行。


18.然后就是Analyze——Macromolecules——open,加载大分子,同样的为了显示好看,可以调整大分子和小分子的模式。

19.然后就是构象分析,我们点击Analyse——conformation——paly,ranked by energy,然后就会出来一个小框框


20.然后我们点击一下这个小框框倒数第二个按钮,我们可以点击Build H-bind——show info,这表示的是第一次对接的结果,然后第一个数值表示的是对接的结合能是-4.43千卡每摩尔,然后蛋白质和这个小分子一共形成了2个氢键,一个就是蛋白质的35号位置的氨基酸,还有一个就是蛋白质的53号氨基酸的位置,这两个氢键的共同的结合能是-4.43,


21.然后我们也可以看其它对接的结果,Analyze——conformation——Load,这样就可以看10个了,然后最后我们可以点击write complex导出我们要的对接的结果,然后需要输入一个PDBQT结尾的文件,一般就输入result.pdbqt。

22.然后我们通过openbabel将这个文件转为PDB文件,这样我们就可以在pymol中打开这个文件了。